More Analysis
The radius of gyration of a protein is a measure of its compactness. If a protein is stably folded, it will likely maintain a relatively steady value of Rg. If a protein unfolds, its Rg will change over time. Let’s analyze the radius of gyration for lysozyme in our simulation:
$ gmx gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
Choose group 1 (Protein) for analysis.
Solution
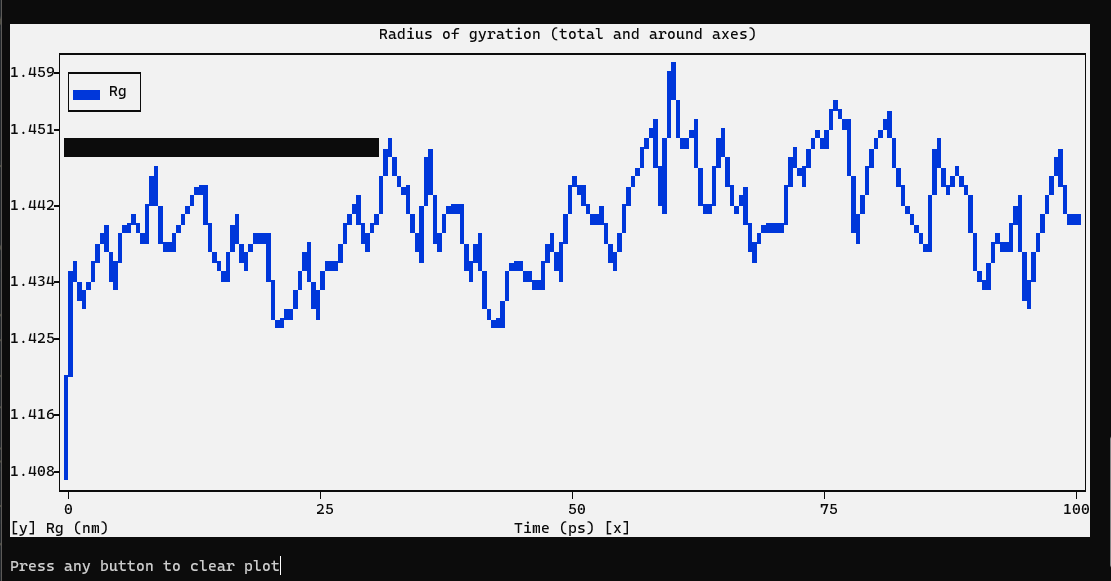
We can see from the reasonably invariant Rg values that the protein remains very stable, in its compact (folded) form over the course of 1 ns at 300 K. This result is not unexpected, but illustrates an advanced capacity of GROMACS analysis that comes built-in.
Summary
You have now conducted a molecular dynamics simulation with GROMACS, and analyzed some of the results. This tutorial should not be viewed as comprehensive. There are many more types of simulations that one can conduct with GROMACS (free energy calculations, non-equilibrium MD, and normal modes analysis, just to name a few). You should also review the literature and the GROMACS manual for adjustments to the .mdp files provided here for efficiency and accuracy purposes.
If you have suggestions for improving this tutorial, if you notice a mistake, or if anything is otherwise unclear, please feel free to email me. Please note: this is not an invitation to email me for GROMACS problems. I do not advertise myself as a private tutor or personal help service. That’s what the gmx-users list is for. I may help you there, but only in the context of providing service to the community as a whole, not just the end user.
Happy simulating!